-

上海沙格企业管理咨询有限公司
主营:MDRCE认证,FDA注册和FDA510K,欧盟自由销售证明,FDA验厂,CE临床评估报告,欧盟授权代表和欧盟注册

上海沙格企业管理咨询有限公司
主营:MDRCE认证,FDA注册和FDA510K,欧盟自由销售证明,FDA验厂,CE临床评估报告,欧盟授权代表和欧盟注册 7
7

口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令辅导)。
EN14683:欧盟医用口罩标准
VFE:过滤效率
PFE:颗粒过滤效率
ASTM F2100:美国医用口罩标准
欧盟标准 EN 14683
有三种测试方法用来给医用口罩进行分类:
1. 体外过滤率(BFE)(ASTM F2101-07)
• 这个测试是用来确定外科口罩上残留的源,此源直接影响到手术室空气中由口罩中释放出的数目。
• 测试方法:使用一个喷雾装置,用来释放出固定流量的空气流,这个空气流包含着一定浓度的金葡萄球菌,让这股空气流穿过包含所有层的外科口罩。喷雾口的平均尺寸为3微米。通过样品的数目和没有样品时的数目进行比较。
• 这个BFE比值越高,说明口罩能更好的保护病人不易因为手术人员而感染。
• BFE => 95% TYPE I
• BFE => 98% TYPE II
• 不同的BFE测试可以称为“修改版的Green&Vesley”或者体外BFE,这种方法在过去经常被使用。这种测试方法能提供较高的值,但并不真正的区分出不同质量外科口罩。这种体外BFE测试方法因此没有成为新的欧盟标准的一部分。
2. 呼吸阻抗 它是指气体在流经呼吸道及呼吸装置时的耗功加总值。
• 这种测试来确定口罩的气流阻力。
• 测试方法:使用一股定量的气流通过包含所有层的外科口罩样品。测量通过样品之前和之后的压力,这个不同的压力值再除以口罩的表面积(cm2)。
• 一个较低水平的呼吸阻抗值说明使用者使用起来更舒服。说明口罩戴起来感觉更加凉爽和更*呼吸,因为在材料上面的压力较小,所以口罩更*保持不变形。这样还能减少从口罩边缘呼出的未经过滤的气体。
分类
• 类别I&II(非防溅)= < 3.0 mmH2O/cm2
• 类别IR&IIR(防溅)= < 5.0 mmH2O/cm2
• 呼吸阻抗通常是用来测量每平方厘米材料上的值。一个增加舒适性的方法就是降低每平方厘米的呼吸阻抗,另外的一个方法就是通过增大口罩的表面积来增大口罩的可过滤的区域。
3. Splash Resistance (ASTM F1862-07)
防溅阻力 指防止血液\体液等溅撒 物的阻止能力.
• 这种测试是用来确定对潜在污染流飞溅的穿透阻碍作用。
• 测试方法:精确定量的人造血液在固定的压力下面通过样品口罩进行喷洒。常用的测试压力一般为80,120和160mmHg。可以通过观察样品口罩背面的是否有通过的液体的痕迹(红颜色)来判断。这个测试在每个压力条件下重复32次,3次以下的液体渗出,就可以判断产品在此压力下具有防溅性能。
• A higher splash resistance means the mask will protect the user in a better way against splashes of potentially contaminated fluid during a surgical procedure.
• 一个较高的防溅性能意味着这个口罩可以更好的保护在手术中使用者不受潜在的污染液体飞溅的侵害。
分类:
• 类别I和类别II不适用
• 类别IR和类别IIR至少为120mmHg
• 120 mmHg是个小的值。这个值和平均的收缩压相一致,用来防止由于小型动脉的破裂造成的小规模的血液飞溅。一些产品的保护级别甚至**过了120mmHg。
依据新的口罩的标准EN14683而定的的性能要求
欧盟标准分类 Bacterial Filtration Efficiency
过滤率 Breathing Resistance
呼吸阻抗
(mmH2O/cm2) Splash Resistance
防溅能力
(mmHg)
I 95% < 3.0 NA
IR 95% < 5.0 120
II 98% < 3.0 NA
IIR 98% < 5.0 120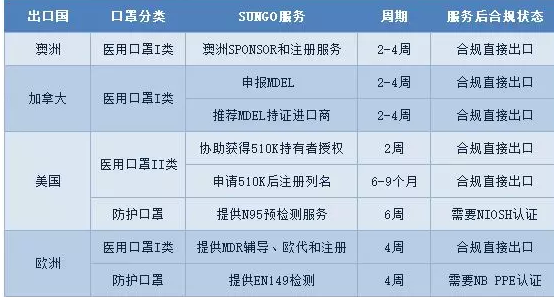
口罩要做的EN14683检测是什么?
我公司专业办理医疗产品出口欧盟、美国、中东南美等国家的各种认证:
TUV莱茵,TUV南德,SGS等CE认证(MDD/MDR法规),全套CE技术文件编订, CE*四版临床评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售证书,ISO13485:二016,医疗器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请),FDA美国代理人服务/ FDA QSR820验厂辅导及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系辅导/OTC验厂辅导及整改,英国BRC认证咨询,BSCI验厂辅导;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令辅导)。
欧盟标准 EN 14683
This standard is intended to help facilitate the choice of surgical face masks in the European Market by standardizing the information and performance data required for the masks.
本标准旨在利用标准化的信息和口罩的性能数据来帮助欧盟市场方便的选择外科口罩。
There are three test methods used to classify surgical masks:
有三种测试方法用来给外科口罩进行分类:
1. Bacterial Filtration Efficiency in vitro (BFE) (ASTM F2101-07)
体外过滤率(BFE)(ASTM F2101-07)
• Test is used to determine the amount of infective agent that is retained by the surgical facemask, which is directly related to the amount of bacteria released through the mask into the air of the surgical theatre.
• 这个测试是用来确定外科口罩上残留的源,此源直接影响到手术室空气中由口罩中释放出的数目。
• To test, a controlled flow of air containing an aerosol with a controlled concentration of Staphylococcus aureas is driven through a sample of the surgical mask containing all layers. The average size of the aerosol droplets is around 3.0 Micron. The number of bacteria that passes the sample is compared to the number that passes without the mask sample.
• 测试方法:使用一个喷雾装置,用来释放出固定流量的空气流,这个空气流包含着一定浓度的金葡萄球菌,让这股空气流穿过包含所有层的外科口罩。喷雾口的平均尺寸为3微米。通过样品的数目和没有样品时的数目进行比较。
• A higher BFE percentage indicates a better protection level for the patient against infective agents from the OR staff.
• 这个BFE比值越高,说明口罩能更好的保护病人不易因为手术人员而感染。
Classification:
分类
• BFE => 95% TYPE I
• BFE => 98% TYPE II
• A different BFE test called the "Modified Green & Vesley" or BFE in vivo has been used frequently in the past. This test method always provides extremely high values, but does not really differentiate between different quality surgical facemasks. The BFE in vivo test method is therefore not part of the new EU standard.
• 不同的BFE测试可以称为“修改版的Green&Vesley”或者体外BFE,这种方法在过去经常被使用。这种测试方法能提供较高的值,但并不真正的区分出不同质量外科口罩。这种体外BFE测试方法因此没有成为新的欧盟标准的一部分。
2. Breathing Resistance (Delta P)
呼吸阻抗 它是指气体在流经呼吸道及呼吸装置时的耗功加总值。
• Test is used to determine the resistance airflow of the facemask.
• 这种测试来确定口罩的气流阻力。
• To test, a controlled flow of air is driven through a sample of the surgical mask containing all layers. The pressure before and after the sample is measured; the difference in pressure is divided by the surface (in cm2) of the sample.
• 测试方法:使用一股定量的气流通过包含所有层的外科口罩样品。测量通过样品之前和之后的压力,这个不同的压力值再除以口罩的表面积(cm2)。
• A lower breathing resistance indicates a better comfort level for the user. It means the mask feels cooler and easier to breath through, and that the mask will maintain its shape in a better way as there is less pressure on the material. There will be less unfiltered air escaping around the mask.
• 一个较低水平的呼吸阻抗值说明使用者使用起来更舒服。说明口罩戴起来感觉更加凉爽和更*呼吸,因为在材料上面的压力较小,所以口罩更*保持不变形。这样还能减少从口罩边缘呼出的未经过滤的气体。
Classification:
分类
• TYPE I & II (non splash resistant) = < 3.0 mmH2O/cm2
• 类别I&II(非防溅)= < 3.0 mmH2O/cm2
• TYPE IR & IIR (splash resistant) = < 5.0 mmH2O/cm2
• 类别IR&IIR(防溅)= < 5.0 mmH2O/cm2
• The breathing resistance is always measured per square centimeter of material. One way to increase comfort is to have a low breathing resistance value per cm2, the other to enlarge the surface of the facemask and thereby the total area available for ventilation.
• 呼吸阻抗通常是用来测量每平方厘米材料上的值。一个增加舒适性的方法就是降低每平方厘米的呼吸阻抗,另外的一个方法就是通过增大口罩的表面积来增大口罩的可过滤的区域。
3. Splash Resistance (ASTM F1862-07)
防溅阻力 指防止血液\体液等溅撒 物的阻止能力.
• Test is used to determine the resistance penetration of potentially contaminated fluid splashes.
• 这种测试是用来确定对潜在污染流飞溅的穿透阻碍作用。
• To test, a precisely determined quantity of specially prepared artificial blood is sprayed at a controlled pressure against a sample of the mask. Frequently these tests are done at pressures of 80, 120 and 160 mmHg. A visible inspection on the backside of the sample indicates if there is a fluid strike-through (red color) or not. The test is repeated 32 times at each pressure, and if three or less masks show a strike through, the product is considered splash resistant at that pressure.
• 测试方法:精确定量的人造血液在固定的压力下面通过样品口罩进行喷洒。常用的测试压力一般为80,120和160mmHg。可以通过观察样品口罩背面的是否有通过的液体的痕迹(红颜色)来判断。这个测试在每个压力条件下重复32次,3次以下的液体渗出,就可以判断产品在此压力下具有防溅性能。
• A higher splash resistance means the mask will protect the user in a better way against splashes of potentially contaminated fluid during a surgical procedure.
• 一个较高的防溅性能意味着这个口罩可以更好的保护在手术中使用者不受潜在的污染液体飞溅的侵害。
Classification:
刚结束的2月后两周,我们接到很多工厂关于医用口罩产品办理国内许可证书的申请。随着国内的口罩供应逐步缓解,同时海外的疫情发展加快,口罩产品出口需求已可以清晰预见。为应对近两天广大客户关于口罩出口认证的问询,SUNGO整理相关国家和地区法规要求以及我们可以提供的服务内容。摘要见表1
Part.1
欧洲市场
欧洲市场对于口罩的管理分为两个主要类别,个人防护口罩和医用口罩。个人防护口罩主要是工业用防护,医用口罩主要是医院使用。
医用口罩
医用口罩对应的欧洲标准是EN14683,该标准对于口罩的分类如下图所示,按照BFE、呼吸阻抗和防喷溅能力分为三个类别,具体指标见表2。
表2 欧洲医用外科口罩的分类
按照医疗器械法规2017/745/EU的要求,口罩产品可以按照一类器械进行管理。依据产品是无菌或非无菌状态提供,其认证模式不一样。
1.非无菌方式提供
1)编制技术文件
2)提供测试报告(可以提供熔喷布性能测试报告和无纺布的生物学报告)
3)编制DOC
4)*欧盟授权代表并完成欧洲注册
2.无菌方式提供
1)灭菌验证
2)建立ISO13485体系
3)编制技术文件
4)提供测试报告(口罩本身的生物学、性能、无菌等测试报告)
5)公告机构审核(目前只能按照MDR审核,预计近期没有NB可以审核)
6)获CE证书
7)*欧盟授权代表并完成欧洲注册
从目前整体情况来看,如果之前没有获得公告机构的CE证书,现在临时去申请已经没有可能性,因此目前出口到欧洲的口罩产品应该只有非无菌状态提供一个选项。但是非无菌并不是对生产环境完全不控制,EN14683对于产品的初始污染菌要求不大于30cfu/g。
2防护口罩
防护口罩的欧洲标准是EN149,按照标准将口罩分为FFP1/FFP2和FFP3三个类别,具体的指标见表3
。
表3 欧洲防护口罩分类
防护口罩需要满足欧盟个人防护设备指令(PPE)的要求,防护口罩属于其中复杂设计的产品。出口欧洲需要授权的公告机构进行认证并颁发证书,认证需要的资料包括:
A)产品的型式试验报告;
B)技术文件评审
C)工厂质量体系审查。
Part.2
美国市场
美国法规对医用口罩和工业防护口罩同样是区分管理,其中医用口罩由FDA管控,而防护口罩则由NIOSH管控。大家熟知的N95口罩就来源于NIOSH对口罩分类中的一个类别。
医用口罩
美国对于医用口罩的管理机构是FDA,在FDA系统中对于口罩的分类代码有如下3个。其中一个是外科口罩,一个是口罩,一个是带有/抗病毒介质的外科口罩,见图1。
三个类别的口罩都属于规则878.4040,分类都是II类,都需要申请510K批准。那么我们正常出口美国的口罩必须的路径为:
1)进行产品测试(性能测试、生物学测试)
2)准备510K文件,提交FDA评审
3)FDA发510K批准信
4)完成工厂注册和器械列名
5)产品出口
但是该过程至少需要半年以上时间。那是否有其他可选方法呢?SUNGO咨询师经过分析提供两种其他可选路径:
(1)已经获得NIOSH批准的N95口罩可以直接注册
从图2可以看出,如果你的N95口罩获得了NIOSH的批准,生物学测试、阻燃测试和血液穿透测试都通过了,那么你是可以豁免510K的,可以直接进行工厂注册和器械列名。
(2)获得持有510K的制造商的授权,作为其代工厂使用其510K批准号进行企业注册和器械列名。申请方需要获得授权书,需要签署正式的质量协议,FDA会进行核实和抽查。如果使用未经许可的号码,将会导致产品召回的风险。
2防护口罩
NIOSH将口罩分成N95、N99、N100、R95、R99、R100、P95、P99、P100合计9个类别。从某种意义上来说,N95算是其中防护级别比较低的品类。
NIOSH的口罩防护等级认证程序复杂,包括:
送样品到NIOSH认可的实验室进行预检,通常是美国实验室;
编写技术文件提交NIOSH评审,通常包括:
✰ 产品图纸
✰ 产品说明
✰ 质量体系文件
✰ 测试报告
工厂检查
核发证书
Part.3澳洲市场
澳洲的医用口罩按照I类管理,需要在TGA进行备案之后可以销售。当然在澳洲的备案需要由澳洲当地的SPONSOR来完成,其合规流程为:
1)*SPONSOR(SUNGO 澳洲可以提供服务)
2)完成技术文档
3)提交TGA进行备案
4)获得证书(见图3)
Part.4
加拿大
加拿大医疗器械产品分为4类,其中1类风险,不需要做器械注册(MDL),只需要进行MDEL(制造商注册),见图4。
图4 加拿大医疗器械管理示意图
加拿大的口罩产品属于I类医疗器械进行管控,按照其法规如果是中国制造商,可以选择:
1)申请MDEL证书自行出口
2)找到加拿大持有MDEL证书的进口商交易
申请MDEL需要完成申请表的如下内容,提交后2-4周可以获得证书。
(1)公司名称及联系方式
(2)许可文件、邮寄和帐单地址
(3)分类和活动表
(4)场地地址
(5)制造商信息
(6)证明
(7)签名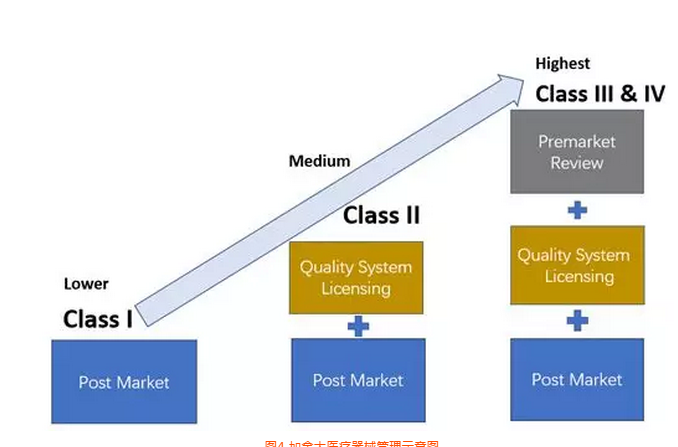
TUV莱茵,TUV南德,SGS等CE认证(MDD/MDR法规),全套CE技术文件编订, CE*四版临床评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售证书,ISO13485:二016,医疗器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请),FDA美国代理人服务/ FDA QSR820验厂辅导及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系辅导/OTC验厂辅导及整改,英国BRC认证咨询,BSCI验厂辅导;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令辅导)。
2017 年4 月5 日,欧洲议会和理事会正式签发了欧盟关于医疗器械*2017/745 号法规(MDR,EU2017/745),5月5日,欧盟期刊(Official Journal of the EuropeanUnion) 正式发布该法规。2017 年5 月25 日,MDR 正式生效, 替代了原医疗器械指令(MDD,93/42/EEC)和主动植入式医疗器械指令(AIMD,90/385/EEC)。
MDR 由指令升级为法规,提高了对欧盟成员国的约束力,具有直接约束性,*各国转化为本国的法律法规的形式即可落实实施。内容上,MDR 在整合原指令的基础上,大幅提升了有关医疗器械认证的规范和限制,例如关于产品分类规则、器械的可追溯性、临床性能研究的规范、增加上市后的产品安全性和有效性的监管等方面。MDR 共10 章123 条,并附有17 个附录。
一、关于法规过渡期
MDR 过渡期为3 年,共涉及四个时间点(见表1):
欧盟医疗器械新法规MDR主要变化情况介绍
仅具有根据90/385/EEC 和93/42/EEC 指令签发的证书的器械可投放市场的前提是自MDR 适用之日起,其在设计和预期目的上无显著变化并符合新法规有关市场后监察、市场监察、警戒、经济运营商及器械注册的规定。
通过豁免指令的形式上市,且符合新法规的器械可在2020 年5 月26 日之前投放市场。并可在2020 年5 月26 日前*并通知符合新法规的符合性评估机构。公告机构可在2020 年5 月26 日前, 采用合规的符合性评估流程并按照新法规签发证书。
对于特定Ⅲ类器械和Ⅱ b 类器械产品,在已委派必要的医疗器械协调小组(MDCG)、*小组前提下,同样可通过指令豁免在2020 年5 月26 日之前投放市场。
法规关于公告机构的要求(正文*35~50 条) 自2017 年11 月26日起适用,即公告机构在新法规发布后的六个月内即应开始进行相应的申请,符合要求后方可依据新法规开展符合性评估。
同时法规对成员国主管机构的*和MDCG 的成立也设定了期限,要求于2017 年11 月26 日前完成。对于成员国主管机构之间的协调,设定期限为2018 年5 月26 日。
二、关于MDR 涵盖产品范围和分类规则
法规对“医疗器械”的定义结合了90/385/EEC 和93/42/EEC 指令的范围,并有所扩大,纳入了有源植入医疗器械、软件以及植入或侵入人体的非医疗用途产品,如隐形眼镜、美容植入物、去除脂肪组织的器械、发射高强度电磁辐射进行皮肤**等操作的器械、利用磁场大脑神经元活动的器械等。对药械组合产品的描述也更为具体,法规还适用于使用活性或非活性动物、人源组织或细胞的产品制造而成的器械。
医疗器械的分类仍延续了之前的大类,即按照风险等级分为四大类:Ⅰ、Ⅱ a、Ⅱ b、Ⅲ类。但分类规则较之前有所增加,由18 条增加至22 条。具体分类规则条款情况及变化情况见下表2:
欧盟医疗器械新法规MDR主要变化情况介绍
三、关于经济运营商各方义务
法规在*I章*2条定义中提出了“经济运营商”的概念,经济运营商是指制造商、授权代表、进口商、经销商以及任何对系统或手术包类器械进行组合或消毒并投放市场的自然人或法人。即在符合法规规定情况下负责器械生产(包括组合或灭菌)、销售及上市后运营的自然人或法人。
法规首先规定了制造商的义务,涵盖生产、合规、上市后监管的产品全生命周期,但法规同时规定,经销商、进口商或其他自然人或法人在向市场提供以其名字、注册商标命名的医疗器械时应承担制造商相应的义务,也包括变更相应器械预期用途或变更其他影响其符合性的事项的情况。在上市后监管要求中,经济运营商同时负有相应的责任和义务。
法规对各方义务的描述更为明确也更为具体,对于制造商的要求更为细化,因此新法规执行后,各方应首先明确自身职责和义务,规范有序地开展生产和市场活动,应审核确认上游供应商是否符合规定,并确认能够证明自己的下游流程符合规定,应按照对应的警戒系统的要求进行或配合事件上报,配合完成现场安全纠正措施,并依据职责组织培训。
四、其他
法规中规定了对于一次性使用器械的再处理即复用的要求。
MDR
*17 条规定,一次性使用的医疗器械的复用只能在相应国家法律允许的情况下进行,且应符合MDR 的规定。任何对一次性使用器械的再处理即复用的自然人或法人应视为复用器械的制造商,承担制造商义务,包括器械的可追溯性。但目前只有部分欧盟成员国接受器械复用并具备相应的法规规定。
MDR 在很多方面的规定都趋于更加严格的监管模式,更加强调持续监管和*协作的监管方式。如从监管层面自上而下确定了欧盟、各成员国、公告机构、经济运营商各自的义务和责任,同时从法规层面设定了成员国之间、公告机构之间及制造商与监管部门之间沟通和协作的制度及途径,从产品监管角度来讲,从产品生产质量体系建立和实施、符合性评估过程中的通用基本要求、技术文件建立、上市后监管文件建立、临床证据等上市前监管要求,到符合性评估程序要求,以及上市后监管、警戒和市场监管等措施,覆盖产品生命周期的全过程,并规定了信息管理的具体要求,包括UDI 及市场监管的电子系统等。
基于本版法规的医疗器械监管将很大程度上提高欧盟对医疗器械产品的要求,不论是制造商还是公告机构都将面临更严格的管理,基于目前的产品分类规则,更多的产品将需要执行公告机构参与的符合性评估流程,更多的品种纳入了医疗器械监管。
五、关于我国审评审批制度可借鉴的思考
基于对法规的研究,在审评审批及监管过程中认为有几点值得借鉴:
首先是整体监管的理念,产品符合性评估程序中不仅包含技术文件审评与生产质量管理体系审核,还包含对上市后监管计划及相关警戒数据报告的审核,且上市后监管责任明确。在产品上市之前即明确了上市后制造商的责任和义务,并使得上市后监管有相应的依据。
其次是持续监管的理念,关于监管评估,法规规定:公告机构应至少每隔12 月开展一次适当的审核和评估,以确保相关制造商采用批准的质量管理体系和上市后监管计划,公告机构至少应每隔五年随机对制造商进行一次现场突击审核。包括对制造商经营场所的审核,必要时还包括对制造商的供应商和/ 或分包商的审核。
另外,上市后临床跟踪的规定,要求制造商主动收集和评估上市后临床数据,旨在确认器械的安全有效性、识别之前未知的并监控已识别的和禁忌症、识别并分析突发风险、确有收益/ 风险的可接受性以及确定器械可能的操作不当或**标示使用以验证其预期用途是否正确。上市后临床跟踪在产品生命周期中的作用不可忽视。
*三,科学监管的理念体现在法规的细节规定或管理要求中。如医疗器械产品种类繁多,法规对于特殊类别产品,其符合性评估程序中分别规定了特殊要求,例如与药物一同使用的器械的认证程序、利用人类或动物源组织或细胞及其物制造器械时的认证程序等,体现特殊产品的个性化要求;对于高风险产品,欧盟法规规定了*小组的职责中包含临床前咨询的程序;对于变更事项的规定,欧盟法规更接近产品本身,如涉及设计或特性的更改、协调标准更改等,使得变更事项更为明确。
另外,欧盟在符合性评估过程中允许产品发生变化,相比于中国法规审评审批过程中无法变更的要求,这种方式更为灵活;充分利用UDI 及电子数据系统等工具, 从欧盟法规层面规定了电子系统的建立和使用要求,从而增加器械上市的透明度和可追溯性。