-

上海沙格企业管理咨询有限公司
主营:MDRCE认证,FDA注册和FDA510K,欧盟自由销售证明,FDA验厂,CE临床评估报告,欧盟授权代表和欧盟注册

上海沙格企业管理咨询有限公司
主营:MDRCE认证,FDA注册和FDA510K,欧盟自由销售证明,FDA验厂,CE临床评估报告,欧盟授权代表和欧盟注册 7
7

欧洲市场对于口罩的管理分为两个主要类别,个人防护口罩和医用口罩。个人防护口罩主要是工业用防护,医用口罩主要是医院使用。
医用口罩
医用口罩对应的欧洲标准是EN14683,该标准对于口罩的分类如下图所示,按照BFE、呼吸阻抗和防喷溅能力分为三个类别。
欧洲医用外科口罩的分类
按照医疗器械法规2017/745/EU的要求,口罩产品可以按照一类器械进行管理。依据产品是无菌或非无菌状态提供,其认证模式不一样。
1.非无菌方式提供
1)编制技术文件
2)提供测试报告(可以提供熔喷布性能测试报告和无纺布的生物学报告)
3)编制DOC
4)*欧盟授权代表并完成欧洲注册
CE认证
2.无菌方式提供
1)灭菌验证
2)建立ISO13485体系
3)编制技术文件
4)提供测试报告(口罩本身的生物学、性能、无菌等测试报告)
5)公告机构审核(目前只能按照MDR审核,预计近期没有NB可以审核)
6)获CE证书
7)*欧盟授权代表并完成欧洲注册
从目前整体情况来看,如果之前没有获得公告机构的CE证书,现在临时去申请已经没有可能性,因此目前出口到欧洲的口罩产品应该只有非无菌状态提供一个选项。但是非无菌并不是对生产环境完全不控制,EN14683对于产品的初始污染菌要求不大于30cfu/g。
防护口罩
防护口罩的欧洲标准是EN149,按照标准将口罩分为FFP1/FFP2和FFP3三个类别。
欧洲防护口罩分类
防护口罩需要满足欧盟个人防护设备指令(PPE)的要求,防护口罩属于其中复杂设计的产品。出口欧洲需要授权的公告机构进行认证并颁发证书,认证需要的资料包括:
A)产品的型式试验报告;
B)技术文件评审;
C)工厂质量体系审查。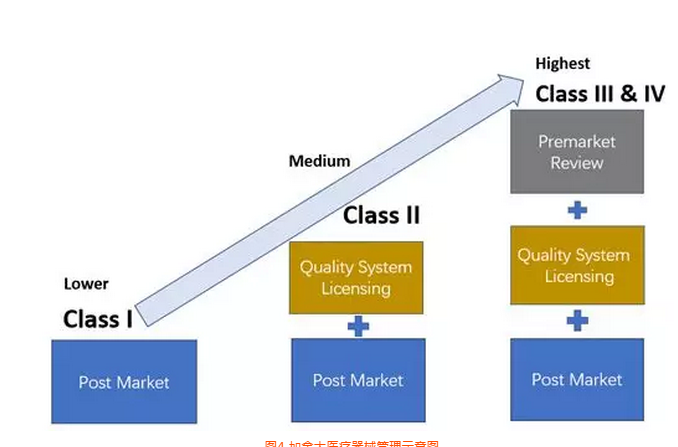
欧盟授权代表和欧盟注册号码是什么?
对欧盟境外的医疗器械制造商来说,无论产品是哪个类别,都需要*一个欧盟境内的授权代表,作为其在欧盟的一个法律实体。
欧盟授权代表(European Authorized Representative )是指由位于欧洲经济区EEA(包括EU与EFTA)境外的制造商明确*的一个自然人或法人。该自然人或法人可代表EEA境外的制造商履行欧盟相关的指令和法律对该制造商所要求的特定的职责。
欧盟授权代表在产品流通在欧盟市场的过程中起到了至关重要的沟通、协调以及解决问题的作用。
在现如今这个信息发达的社会,很多制造商都知道欧盟授权代表的定义,以及欧盟授权代表主要承担了哪些职责。
那具体细节如何执行,有哪些特殊的要求等问题,可能很多医疗器械制造商并不是十分了解。我们参考了欧盟具体的指南文件,从几个问题中介绍一下法规的具体要求:
1. 哪些技术文档必须要发给欧盟授权代表处进行保存?
授权代表有义务保留国家主管部门掌握的某些信息,例如合格声明和技术文件(AIMDD附件2*6.1节; MDD附件II*6.1节,附件III*7.3节,附件IV*7节,附件V部分) 5.1,附件VI*5.1节,附件VII*2节; IVDD*9(7)和10(3)条)。
授权代表必须能够提供市场监督机构为进行市场监督而可能需要的所有文件和信息(*765/2008 / EC号条例*19条)。
当局根据转换指令或根据*765/2008 / EC号条例的国家立法提出任何信息请求。因此,关于这种请求或“命令”的合法性与否的任何问题都是由国家法院决定的。
该信息可以与授权代表一起存储,授权代表应被授权将信息直接分发给当局。在这种情况下,合同应包括制造商的义务,以保持信息始终更新。
如果制造商选择不向授权代表存储信息,他应向授权代表提供市场监督机构在接收到授权代表转发的请求后可能需要的所有文件和信息,以便进行市场监督。制造商、授权代表应该可以访问所有文档和信息。
在这种情况下,合同必须确保制造商及时向授权代表提供所要求的信息,并且应包括制造商的义务,以便随时向授权代表通报任何变更。授权代表如果后者未向他提供获取必要信息的,则应撤销与制造商的合同。
很多客户不愿意把产品技术文档交给欧代,或者会提供部分的产品技术文档,按照欧盟针对欧盟授权代表的指南文件MEDDEV2.5-10要求,欧代必须要保留至少以下的文件:
i) Declaration of conformity,
ii) Copy of the label, packaging and instructions for use (in all languages requested by the countries where the device is marketed),
iii) Notified Body certification (where relevant),
iv) Post market surveillance process and data, vigilance reports and complaints, processes and data
v) Technical documentation relevant to market surveillance investigation being undertaken by the Member State,
vi) Relevant clinical data / notification,
vii) Details of any distributors / suppliers putting the CE marked devices on the market,
viii) Incident reports and corrective actions taken.
2. 产品出口到欧盟前,欧代是否必须要把制造商的信息和产品的信息向所在国注册?
按照欧盟93/42/EEC MDD的要求*14条的要求,I类的产品在出口到欧盟前必须要由其欧盟授权代表向其所在国进行通报/注册。但是像德国,除了MDD指令外德国也有单独的德国医疗器械法规MPG,MPG要求德国的欧盟授权代表要将所有类别医疗器械的信息在次出口到欧盟前向其主管当局进行通报注册。
所以,选择不同的国家的欧盟授权代表也会造成要求的不同,例如选择英国欧盟授权代表的话,I类产品必须在MHRA进行注册,IIA,IIB,III类产品无法进行注册。选择德国欧盟授权代表的话,所有类别产品都必须在出口到欧盟前向DIMDI注册。
3. 是否可以选择多个欧盟授权代表?
按照欧盟授权代表的指南文件MEDDEV2.5-10要求,一个公司可以有多个授权代表,但是同一个产品有且只能选择一个欧盟授权代表。
很多制造商对这块的要求不是很明确,管理上也不精细,很随意的就*一个欧盟授权代表,却不知这样回头会造成很大的隐患,如果产品将来在欧盟出现了事故,欧盟主管当局将不知道联系谁,会造成事故处理的不及时或严重滞后,也会给主管当局在处理事故时造成一个很不正规的印象。
4.简言之
为了更好地保护欧盟的消费者和环境,欧盟的法律要求,为了实现产品的可追溯性,制造商投放到欧盟市场的加贴了CE标志的产品必须标有制造商的名称和联络地址;如果制造商来自欧洲经济区EEA(包括EU与EFTA)以外的国家,其产品必须同时标有制造商和制造商的欧盟授权代表的名称和联络地址。
欧盟授权代表的职责包括
1/ 作为制造商*的授权代表,负责与欧盟范围内各个国家的医疗器械监管机构联系,处理医疗器械的事故、投诉、不良事件以及召回等工作;
2/ 保留制造商的CE技术文件,当监管机构提出问题时,进行联络制造商、回复和沟通;
3/ 受制造商的委托,在欧盟进行医疗器械产品注册;
4/ 受制造商的委托,申请欧盟颁发的自由销售证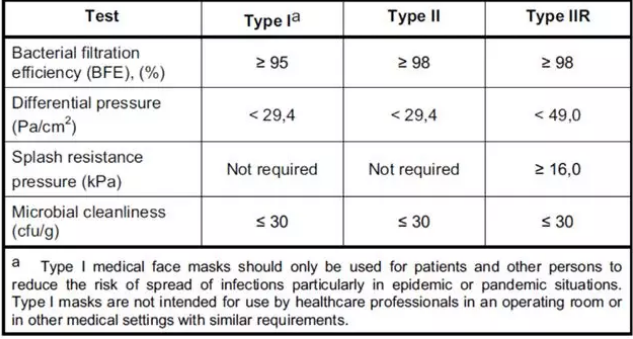
公司从2017年年初开展临床评估业务以来,为国内近两百家医疗器械企业提供了临床评估报告编撰服务,其中包括很多国内**的大型医疗器械企业。*四版欧盟临床评估指南的发布,的确给企业带来了不小的挑战,此文对主要变化进行了梳理。
主要变化之一:临床报告更新的频率
按照新版临床报告指南的要求,对于高风险或者新设备,应每年更新;对于低风险的设备,每2-5年更新。对于如何确定更新的额频率需要有定义。对于任何风险类别的器械,如果从PMS收集到的信息影响到评价或者结论,CER需要进行更新。
主要变化之二:报告编写人和评价人的
按照新版临床报告指南的要求,对于临床报告的编写人提出了要求。包括需要有相关专业的高等教育学位以及至少五年的专业经历,或者十年的专业工作经历,如果学位不够的话。如果不能满足要求,需要对其进行判定和说明。
主要变化之三:评价报告需要有明确的可测量的目标
*四版临床评估指南对于临床评估报告的目的有更明确的描述,需要与器械的安全性、性能以及风险-收益平衡进行更加清晰和详细的描述,在指南的*7部分和附件5中有详细的描述。
主要变化之四:确定技术发展水平
*四版临床评估指南对于设备的技术发展水平和处理方式的建立和文件化,提供了更加详细的描述。这包括确定设备的安全性和性能,以及被宣称的比对器械,行业的基准器械,或者其他的类似器械;需要包括风险和收益的分析。
主要变化之五:数据的科学性和有效性
*四版临床评估指南非常强调数据的科学性和有效性,包括从统计学考虑。这贯穿于整个指南文件规定的阶段,包括影响数据完整性的因素,数据的客观性和权重,文件搜集的方法,数据的评估和权重,数据是否阐述了符合性的分析。
主要变化之六:比对器械
*四版临床评估指南对于评估器械和比对器械的等效有了更加详细的规定,特别强调对于比对器械的数据的可获得性。需要从临床数据、技术参数、生物性能三个方面逐一比对,确保所有比对的内容不存在差异。
主要变化之七:比对器械的数据获得
*四版临床评估指南要求公告机构对于企业是否能够获得比对器械的数据进行挑战,这个被认为是法规的一个转折点,这要求制造商需要有一个被允许接触竞争对手的器械数据的协议。
主要变化之八:什么时候需要临床试验
*四版临床评估指南附件2详细描述了器械的风险以及制造商怎么决定是否具备了充分的临床证据。
主要变化之九:风险-收益
附件7提供了详细的指南,对于器械的安全性和性能表述;
附件7.2 讨论了风险和收益分析,包括对于风险和收益的量的评估,以及总体评价。交付后的数据价值,以及可能会影响统计有效性行的因素等。
主要变化之十:售后监督和售后临床跟踪
*四版指南强调了临床评估、售后监督和售后临床评估的关系。附录12强调了公告机构需要确认PMCF被很好的策划了,以及依据所收到的数据来描述其满足CER。
近年来SUNGO为国内众多制造商提供了FDA验厂的辅导和翻译陪审服务。这其中包括了FDA提**天通知验厂的时间较端紧迫的案例,也包括了为连续两次验厂失败的制造商解除警告信并移除进口禁令的复杂度较高的案例。
PART 1
FDA工厂审查的概况
FDA每年会对**的医疗器械制造商进行抽样审查,作为其进行售后市场监管的主要途径之一。所有的审查都会由美国FDA的工作人员进行,不论这些人是什么族裔,他们都是美国籍,都代表了美国的利益。
近几年,在美国以外的国际市场,中国制造商的被抽样量一直稳居****。目前中国在FDA的注册制造商约为4500家左右,每年抽查的概率在2-3%。通常FDA工厂审查会由1名审查官进行为期4天的现场审查。制造商*支付任何审查费用。
PART 2
FDA工厂审查的直接后果
大部分的中国制造商收到美国FDA的审查通知都会比较重视,基本上都会积极应对,动员内外部的力量和资源来确保审查顺利进行。当然也有部分制造商不了解审查可能会导致的结果,没有给予足够的重视,导致后面很被动的局面。
FDA工厂审查的直接结果会有三种,分别是NAI,VAI和OAI。
代码 全称 中文译意
NAI No Action Indicated *采取整改
VAI Voluntary Action Indicated 自愿采取整改
OAI Official Action Indicated 强制采取整改
NAI表示在FDA工厂审查时,没有开出任何书面形式的不符合项(由于FDA的不合格报告表单的编号为483,FDA也将不符合简称“483”),也可以称为“零483”。
VAI表示在FDA工厂审查时,FDA审查官发现了工厂的管理系统有违背FDA的质量体系法规的内容,进而开具了书面形式的不符合项,也可以开具了“483”。“483”的个数可能是1个,也可能是20个或更多。只要工厂按照FDA的要求积极整改,提供充分的证据,都不会导致更多后果。
OAI表示在FDA工厂审查时,FDA审查官发现了工厂管理系统存在严重违背FDA的质量体系法规的内容,或者是没有能够按照FDA的要求对于VAI进行及时充分的整改,而开具的警告信(Warning Letter)。如果仅仅是开具了警告信而没有上Import Alert,制造商的产品依然可以出口,但是警告信会公布在FDA网站上,会影响美国客户对制造商的信心,必须尽快采取措施解除。
PART 3
FDA工厂审查的后果放大路径
FDA的审查之所以让很多制造商觉得紧张,是因为稍有不慎,其结果的严重性可能会*放大,终让制造商失去整个美国市场。其放大路径如下:
1. VAI没有按照FDA要求进行充分整改,会发展成为OAI;
2. OAI没有按照FDA要求及时响应,会被列入Import Alert;
3. 进入了Import Alert,企业的出口产品入境时会被自动没收(DWPE)。
由于FDA工厂审查导致的Import Alert要移除,通常都需要进行再次的工厂审查。除了再次的工厂审查之外,还有海量的证据需要随同书提交FDA审查。从开始处理到终完成Import Alert移除的过程,效的处理周期需要一年时间。当然伴随着的还有巨额的费用。
SUNGO在2016年帮助某中国制造商(其在2013年和2015年两次验厂失败,背有两封警告信和进口禁令)成功解除了警告信并移除进口禁令。问题能在一年内被我们解决,项目算是很顺利,但是这家制造商丢掉了原有的大部分的美国市场,损失很大。
值得一提的是,很多国内的制造商,碰到产品不能出口时通常会寻求国际律所的帮助,例如本杰明、霍根路伟等。但实践表明,技术法规的符合性问题大部分时候并非是律所可以解决的。
PART 4
避免严重后果的方法
面对可能的严重后果,预防永远是有效和成本的途径。采取预防措施,我们的建议是:
1) 当您的产品进入美国市场之前,尽快建立起QSR820 体系。
2) 寻求第三方的专业机构进行辅导。
3) 寻求第三方的专业机构进行模拟审核。
4) 在收到FDA验厂通知时,尽快联系专业机构提供支持。
特别指出的是:有部分企业终被开具警告信并进入Import Alert是由于FDA现场审查时候的翻译人员对于技术法规和公司的质量管理系统不清楚导致翻译不准确,终导致审查员开具了很多不应该有的不符合。因此,寻找专业的翻译陪审也非常关键。
SUNGO的FDA验厂辅导优势 :
1) 辅导团队具备多年**美资企业的法规工作经验;
2) 具有成功辅导包括新华医疗集团在内的近百家企业通过FDA审核的经验;
3) 具有成功辅导FDA提**天通知的飞行检查的经验; 咨询师具有优秀的语言能力,担任陪审和翻译的任务会使得企业应对更轻松。